


Talassemia
La Talassemia racchiude un ampio gruppo di anemie croniche dovute a ridotta o assente produzione di emoglobina, cioè di quella proteina che si trova all’interno dei globuli rossi del sangue ed è deputata al trasporto dell’ossigeno a tutti i tessuti dell’organismo.
In base alla gravità del quadro clinico ed alla necessità di supporto trasfusionale continuo le talassemie vengono distinte in due grandi gruppi:
• le Talassemie Trasfusioni Dipendenti (TDT)
• le Talassemie Non Trasfusione Dipendenti (NTDT)
Entrambe le forme sono causate dalla presenza di mutazioni sui geni che codificano per le catene dell’emoglobina, ereditate generalmente dai due genitori portatori sani della malattia (che hanno solo un gene mutato).
Le mutazioni sui geni dell’emoglobina sono molto frequenti nei soggetti provenienti da Paesi che si affacciano sul Mar Mediterraneo, in Medio Oriente, in Asia e in India ma, in seguito ai flussi migratori e all’aumento di coppie con etnia mista, attualmente soggetti portatori e affetti da talassemia sono presenti in tutto il mondo.
Talassemia trasfusione dipendente (TDT)
La Talassemia Trasfusione Dipendente, conosciuta anche come Talassemia Major o Morbo di Cooley, è un’anemia congenita che necessita:
• di trasfusioni regolari e continuative dai primi mesi di vita per prevenire le complicanze legate al valore basso di emoglobina
• di una terapia ferrochelante in grado di eliminare il ferro in eccesso che inevitabilmente si accumula con le trasfusioni e che, a livelli elevati nell’organismo, è estremamente tossico.
Attualmente i pazienti che eseguono un regime trasfusionale regolare, mantenendo un adeguato livello di emoglobina pre-trasfusionale ed una corretta terapia ferrochelante con un buon controllo dei livelli di ferro dell’organismo, hanno aspettativa e qualità di vita elevata.
Come si manifesta?
La talassemia trasfusione dipendente è caratterizzata da anemia (bassi valori di emoglobina) causata sia da alterata produzione di globuli rossi nel midollo osseo (eritropoiesi inefficace), sia dalla loro prematura distruzione, cioè emolisi. I primi sintomi di malattia si sviluppano tra i 6 e i 24-36 mesi e spesso sono aspecifici con pallore, ittero (cioè colorito giallastro della cute o delle sclere), riduzione dell’alimentazione e riduzione o arresto della crescita. Data l’anemia cronica, il midollo osseo tende a supplire aumentando la produzione, ma la sua espansione eccessiva, induce negli anni deformità ossee, soprattutto a livello del cranio e del volto. L’anemia grave e prolungata determina scarso accrescimento ponderale e talvolta sviluppo di problematiche cardiache. La milza, organo deputato alla distruzione dei globuli rossi anomali dal circolo sanguigno periferico, si ingrossa anche notevolmente (splenomegalia).
Come si diagnostica?
La diagnosi di sospetto è possibile con un esame emocromocitometrico e con l’elettroforesi dell’emoglobina. L’analisi genetica delle catene alfa e beta globiniche confermerà la presenza di mutazioni causative di malattia.
Lo stesso emocromo ed elettroforesi dell’emoglobina nei genitori mostreranno in entrambi lieve anemia microcitica e lo stato di portatore di tratto talassemico o anemia mediterranea.
Come si prevengono le complicanze?
La prevenzione delle principali complicanze della talassemia è basata su un programma multidisciplinare di sorveglianza attiva che il centro unitamente alla famiglia mette in atto con controlli periodici clinici edesami ematochimici; a questi si associano, generalmente a cadenza annuale, esami strumentali:
• ecografia dell’addome,
• risonanza magnetica T2* cuore e fegato,
• elettrocardiogramma, ecocardiogramma e visita cardiologica,
• visita oculistica ed ortottica.
Vi sono dei controlli che sono indicati in alcuni periodi della vita o con frequenza differente:
• visita otorinolaringoiatra e audiometria,
• studio del metabolismo dell’osso e mineralometria ossea,
• valutazione endocrinologica.
E’ anche raccomandato un supporto psicologico per il bambino e la sua famiglia.
Il soggetto con talassemia viene inserito dal medico specialista presso il centro di riferimento regionale, nel Registro Malattie Rare ed ha diritto ad una esenzione ticket, contraddistinta dal codice RDG010.
Come si cura?
Oltre a terapia di supporto vitaminica con acido folico e vitamina D, il trattamento della talassemia trasfusione dipendente si basa su due pilastri fondamentali:
– Trasfusioni croniche
Per prevenire le complicanze legate all’anemia e all’eritropoiesi inefficace sono necessarie trasfusioni periodiche di globuli rossi concentrati. L’obiettivo del regime trasfusionale cronico è mantenere un valore di emoglobina pre-trasfusionale tra 9 e 10 g/dL per assicurare una crescita ottimale, sopprimere adeguatamente l’iperattività del midollo, evitare le deformità ossee e mantenere una buona attività psicofisica.
Le trasfusioni vengono eseguite mediamente ogni 15-21 giorni, ma l’intervallo trasfusionale può variare in base ad eventi contingenti (febbre o infezioni concomitanti, quantità di sangue trasfuso, etc.).
Il regime trasfusionale continuativo se da un lato cura le complicanze legate all’anemia, dall’altro determina accumulo di ferro nel fegato, nelle ghiandole endocrine e nel cuore, causando negli anni disfunzione di tutti questi organi. Infatti se il ferro è un elemento fondamentale per l’attività di tutte le cellule dell’organismo, quando raggiunge livelli elevati diventa estremamente tossico. Il sovraccarico di ferro è inevitabile in chi riceve trasfusioni regolari, in quanto l’organismo umano non è in grado di eliminare il ferro in eccesso.
– Ferrochelazione
Per prevenire o trattare le complicanze causate dal ferro, è fondamentale la terapia ferrochelante, cioè farmaci in grado di legare ed eliminare il ferro patologico in eccesso. Al momento sono disponibili i seguenti farmaci ferrochelanti:
• il deferasirox, disponibile in compresse, si assume per bocca una volta al giorno, generalmente al mattino. È raccomandata una abbondante idratazione giornaliera. In caso di febbre, vomito o diarrea il farmaco viene interrotto per qualche giorno.
• il deferiprone,disponibile in compresse e sciroppo, viene assunto per bocca 3 volte al giorno. E’ previsto il monitoraggio dell’emocromo nel primo anno di assunzione del farmaco per il possibile calo dei globuli bianchi che talvolta si può riscontrare. Il farmaco va interrotto prontamente in caso di febbre per essere ripreso a distanza di 48 ore dallo sfebbramento.
• la deferoxamina, somministrata in infusione sottocutanea continua per 10-12 ore, di notte, attraverso una pompa infusiva ed un deflussore che terminano conun ago sottocutaneo da applicarsi mediante apposito cerotto su addome, braccia, cosce variando costantemente il sito di infusione. Raramente, per ottenere una rimozione ottimale del ferro (ad esempio in preparazione al trapianto di midollo osseo), può rendersi necessaria una infusione continuativa del farmaco per 24 ore ed in tal caso è necessario posizionare un catetere venoso centrale (cioè un accesso venoso fisso con un tubicino che aggetta all’esterno del corpo). La medicazione del catetere viene eseguita in ospedale settimanalmente, usualmente in concomitanza alla ricarica del farmaco nel serbatoio della pompa.
Sono in fase di studio, anche in età pediatrica, nuovi farmaci chelanti orali.
La terapia ferrochelante, indipendentemente dal tipo di farmaco utilizzato, va assunta continuativamente e costantemente, ogni giorno, con regolarità, attenendosi alle indicazioni mediche. Ciò permette di prevenire lo sviluppo di sovraccarico marziale significativo a livello epatico, di evitare il sovraccarico cardiaco ed endocrino, ridurre il rischio di complicanze di malattia.
Come si monitora il sovraccarico di ferro?
Il sovraccarico di ferro può essere sorvegliato attraverso:
• ferritina: proteina di deposito del ferro, il suo aumento persistente indica livelli patologici di ferro dell’organismo. Aumenta però anche in corso di infiammazione o di infezione e pertanto è un indice non preciso del sovraccarico. Viene determinata attraverso un prelievo del sangue.
• risonanza magnetica epatica e cardiaca: tecnica che offre una determinazione precisa dei depositi di ferro nell’organismo. È necessario monitorarla annualmente in chi riceve trasfusioni croniche continuative. Può essere eseguita però in bambini dai cinque-sei anni di vita e richiede talvolta adeguata preparazione all’esame perché richiede di mantenere la posizione ferma a lungo, in un ambiente, quella della risonanza, angusto e rumoroso.
• SQUID o biosuscettometria: metodica non invasiva che fornisce una stima realistica del quantitativo di ferro nel fegato. Questo macchinario è disponibile solo presso il centro di Torino. Può essere eseguito anche in bambini piccoli, dopo i tre anni di vita.
• biopsia epatica: tecnica che offre la valutazione più precisa, reale ed accurata del sovraccarico di ferro. Data l’invasività della procedura, il suo utilizzo oggi, ancor più in età pediatrica, è estremamente limitato a casi specifici.
– Nuovi farmaci
Diverse sono le nuove molecole in fase di sperimentazione per la talassemia. Il trattamento di questa patologia nei prossimi anni vedrà certamente cambiamenti significativi con la possibilità di impiego di composizioni di farmaci e personalizzazione della terapia.
Nel novembre 2021 è stato approvato da AIFA (Associazione Italiana Farmaci) un nuovo farmaco per il trattamento della talassemia trasfusione dipendente: il luspatercept si è dimostrato capace di migliorare l’eritropoiesi inefficace, riducendo, in una percentuale significativa di pazienti, la richiesta trasfusionale. Tale farmaco è al momento disponibile nell’adulto (sopra i 18 anni), ma è in sperimentazione anche in età pediatrica con risultati altrettanto promettenti. L’infusione, sottocute, viene eseguita in ambiente ospedaliero ogni tre settimane.
Esistono terapie curative della talassemia?
– Trapianto di midollo osseo
Il trapianto di midollo osseo permette di curare la talassemia trasfusione dipendente. Negli ultimi anni, grazie al miglioramento delle tecniche trapiantologiche, dei farmaci utilizzati e delle terapie di supporto, la sopravvivenza libera da malattia si raggiunge in circa 80-90% dei casi.
L’epoca migliore per eseguire il trapianto di midollo, è la prima infanzia, l’età prescolare o comunque entro i dieci anni di vita.
Il trapianto è possibile in presenza di un donatore familiare HLA identico (cioè con lo stesso codice genetico: ogni fratello ha il 25% di possibilità di essere HLA identico) oppure di un donatore da banca compatibile (cioè con una alta compatibilità del sistema HLA). Per definizione il genitore dona al proprio figlio metà del proprio corredo genetico e pertanto la compatibilità HLA è del 50%. Trapianti aploidentici, cioè da genitore, vengono eseguiti con buoni risultati.
Le principali complicanze del trapianto di midollo osseo sono:
• le infezioni: dovute all’immunosoppressione indotta dalla chemioterapia di preparazione al trapianto, cioè il regime di condizionamento; la depressione del sistema immune perdura per diversi mesi per risolversi progressivamente allontanandosi dal trapianto;
• il rigetto: dovuto al ricevente che rifiuta le cellule del donatore. Piu’ spesso si tratta di rigetto secondario, cioè che avviene dopo l’attecchimento del midollo osseo. Nelle settimane/mesi seguenti, lentamente, la quota di midollo appartenente al donatore (chimerismo) cala progressivamente. Il bambino sta bene, ma può ritrovarsi a distanza dal trapianto nuovamente talassemico per la ricostituzione del proprio midollo osseo;
• la malattia trapianto verso l’ospite o graft o GVHD (cioè graft-versus-hostdisease): sono le cellule del donatore che non riconoscono il corpo del ricevente e lo attaccano. È un processo immune che può coinvolgere i diversi organi del corpo e che può durare mesi. Richiede il trattamento con farmaci immunosoppressori.
– La Terapia Genica
La terapia genica è la nuova frontiera verso la cura definitiva della talassemia trasfusione dipendente. Questo approccio terapeutico consiste nell’inserire il gene correttamente funzionante nelle cellule midollari dei pazienti talassemici. Si tratta di un trapianto autologo di cellule staminali con rischi e complicanze inferiori rispetto al trapianto di midollo (assente rischio di rigetto e di malattia trapianto verso l’ospite, inferiori i rischi infettivi). Al momento è una terapia non più sperimentale, che ha dimostrato non solo la sua sicurezza ma anche l’efficacia, che attende l’avvallo da parte degli organi competenti internazionali e nazionali per l’impiego.
Talassemia non trasfusione dipendente (NTDT)
La Talassemia Non Trasfusione Dipendente o Talassemia Intermedia, è un’anemia congenita (cioè acquisita dalla nascita), cronica che non necessita di trasfusioni regolari e continuative. Trasfusioni occasionali possono essere necessarie per gestire eventi acuti che causano una riduzione improvvisa del valore di emoglobina.
L’aspettativa e la qualità di vita sono molto elevate se vengono eseguiti i controlli necessari ad identificare precocemente le complicanze della malattia. I controlli periodici, la cui frequenza viene stabilita dal centro di riferimento e può cambiare in base alle diverse età, riguardano:
• esami ematochimici: emocromo, funzionalità epatica e renale, indici di emolisi, esami ormonali;
• esami strumentali: elettrocardiogramma ed ecocardiogramma, ecografia dell’addome, visita oculistica, RMN T2* epatica, mineralometria ossea.
Come si manifesta?
La talassemia non trasfusione dipendente si caratterizza sempre per anemia cronica (bassi valori di emoglobina) che può essere lieve o moderata e non richiede trasfusioni regolari associata a ittero cutaneo, cioè colorito giallastro della cute e delle sclere, di entità variabile.
Quali sono le complicanze?
Anche i soggetti che non ricevono trasfusioni regolari, possono sviluppare sovraccarico di ferro, soprattutto nel fegato e nelle ghiandole endocrine, a causa di un eccessivo assorbimento del ferro a livello intestinale. Il ferro tossico, negli anni può portare, se non monitorato ed eventualmente trattato, a patologie del fegato ed endocrine (malattie della tiroide, ritardo della pubertà, riduzione della fertilità etc.).
Una complicanza dell’adulto è il rischio di trombosi presente soprattutto nei soggetti splenectomizzati, cioè a cui è stata asportazione chirurgicamente la milza. Per prevenire le trombosi può essere indicato un trattamento con farmaci anticoagulanti o antiaggreganti, come l’aspirina a basse dosi; raramente, nei casi ad alto rischio o con storia di trombosi pregresse, si può rendere necessario il regime trasfusionale cronico. Questo rischio, insieme a quello di infezioni gravi, è uno dei motivi per cui oggi la splenectomia non è più un trattamento di routine nella talassemia, ma va riservata a casi molto selezionati.
Negli anni la distruzione cronica di globuli rossi, cioè l’emolisi cronica, può portare allo sviluppo di calcoli alla colecisti che possono rendersi sintomatici con coliche biliari o causare problematiche infettive (colecistite) oppure ostruttive epatiche come l’ittero colestatico per il blocco di un calcolo nelle vie biliari. La presenza di calcoli biliari viene individuata con l’ecografia dell’addome e può condizionare l’indicazione alla rimozione chirurgica della colecisti (colecistectomia).
Raramente il tessuto eritropoietico (che produce globuli rossi) al di fuori del midollo osseo, eritropoiesi extramidollare, può proliferare in maniera incontrollata a livello paraspinale (a fianco alla colonna vertebrale) con il rischio di compressione delle fibre nervose e causando disturbi neurologici importanti.
Come si diagnostica?
Come per la Talassemia Trasfusione Dipendente, anche questa forma di talassemia si sospetta mediante l’emocromo e l’elettroforesi dell’emoglobina anche se la conferma diagnostica è posta dall’analisi molecolare che identifica le due mutazioni che causano la malattia.
Come si cura?
L’anemia cronica può richiedere trasfusioni di emoderivati occasionali nelle seguenti condizioni:
• infezioni,
• interventi chirurgici,
• gravidanza.
Indipendentemente dal valore di emoglobina, un regime trasfusionale regolare può essere indicato quando:
• riduzione o arresto della crescita
• ritardo dello sviluppo puberale,
• ridotta tolleranza all’esercizio fisico, deformità ossee, riduzione del livello di emoglobina associato ad un incremento progressivo delle dimensioni della milza, scarsa qualità di vita.
Il regime trasfusionale apporta una quantità eccessiva di ferro all’organismo che deve essere eliminato con farmaci ferrochelanti (vedi il paragrafo sulla Ferrochelazione e sul controllo del sovraccarico di ferro nella talassemia trasfusione dipendente).
Nuovi farmaci, come il luspaterceptin grado di migliorare l’eritropiesi inefficace, cioè riducendo la quota di globuli rossi distrutti prematuramente, sono in fase avanzata di sperimentazione con risultati molto incoraggianti in termini di aumento del valore di emoglobina medio e riduzione delle possibili complicanze.
Il portatore sano di Talassemia o di Anemia Mediterranea
o di altra variante emoglobinica
La forma più conosciuta di anemia mediterranea è lo stato di portatore dibeta-talassemia. Il soggetto portatore sano di beta talassemia presenta generalmente una microcitosi (cioè dimensioni dei globuli rossi ridotte) associata o meno a lieve anemia (cioè leve riduzione dei livelli di emoglobina) che non condiziona alcuna sintomatologia.
La trasmissione è autosomica recessiva, cioè un portatore eredita dal proprio genitore tale condizione e la trasmette nel 50% dei casi ad ogni gravidanza ai propri figli.
In passato i portatori di anemia mediterranea erano diffusi prevalentemente nei Paesi che si affacciano sul Mar Mediterraneo, in Medio Oriente, in Asia e in India ma, in seguito ai flussi migratori, attualmente soggetti portatori sono presenti in tutto il mondo.
La diagnosi si basa sull’emocromo e sull’elettroforesi dell’emoglobina.
Non sono richiesti né esami periodici, né terapia specifica (può essere talvolta utile una integrazione vitaminica con acido folico).
Lo stato meno noto di portatore di anemia mediterranea è quello di portatore dialfa talassemia che può essere distinto in alfa talassemia silente e in tratto alfa-talassemico a seconda che siano carenti uno oppure due dei quattro geni codificanti per le alfa globine (soggetto normale: αα/αα; alfa talassemia silente α-/αα; tratto alfa-talassemico α-/α- oppure –/αα). IL portatore sano di alfa talassemia presenta solo una microcitosi, raramente lieve anemia.
La diagnosi di presunzione si basa sull’emocromo e sull’elettroforesi dell’emoglobina, ma la conferma diagnostica, prescritta dal medico genetista in caso di rischio riproduttivo della coppia genitoriale (cioè nel caso in cui entrambi i genitori sono portatori), si ha con i test molecolari.
Alcuni soggetti sono portatori sani divarianti anomale dell’emoglobina. Ne esistono diverse (ad esempio emoglobina C, emoglobina D, emoglobina E, etc.): alcune causano microcitosi dei globuli rossi, talvolta associata a lieve anemia, mentre l’identificazione di altre avviene solo per nota familiarità o del tutto casualmente come riscontro occasionale.
La diagnosi avviene sempre mediante l’emocromo e l’elettroforesi dell’emoglobina.
Se due partner sono entrambi portatori di anemia mediterraneo o di variante emoglobinica presentano fino al 25% di possibilità di avere figli malati di talassemia o patologia dell’emoglobina.
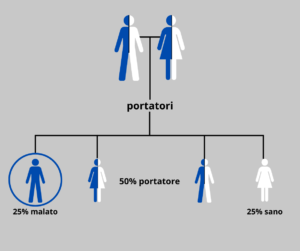
Consulenza genetica e diagnosi prenatale
La consulenza genetica deve essere eseguita nelle coppie a rischio riproduttivo in cui entrambi i genitori sono portatori sani di anemia mediterranea (alfa o beta talassemia) o di altra variante emoglobinica (anche il portatore sano di altre varianti emoglobiniche, combinandosi con il portatore di alfa o beta talassemia può trasmettere la talassemia o altra anemia cronica alla prole).
Gli esami del sangue basilari che il partner di un portatore sano deve eseguire sono l’emocromo e l’elettroforesi dell’emoglobina (il solo emocromo “riferito nella norma” NON è sufficiente ad escludere il rischio!).
La consulenza genetica fornisce le informazioni necessarie, permette la conferma diagnostica molecolare con l’identificazione delle mutazioni ed eventualmente avvia alla diagnosi prenatale. Con le mutazioni genetiche identificate nei genitori è possibile eseguire la villocentesi e le conseguenti decisioni del caso.